
A small-molecule drug is the first to preserve hearing in a mouse model of an inherited form of progressive human deafness, report investigators at the University of Iowa, Iowa City, and the National Institutes of Health’s National Institute on Deafness and Other Communication Disorders (NIDCD). The study, which appears online in Cell, sheds light on the molecular mechanism that underlies a form of deafness (DFNA27), and suggests a new treatment strategy.
“We were able to partially restore hearing, especially at lower frequencies, and save some sensory hair cells,” said Thomas B. Friedman, Ph.D., chief of the Laboratory of Human Molecular Genetics at the NIDCD, and a coauthor of the study. “If additional studies show that small-molecule-based drugs are effective in treating DFNA27 deafness in people, it’s possible that using similar approaches might work for other inherited forms of progressive hearing loss.”
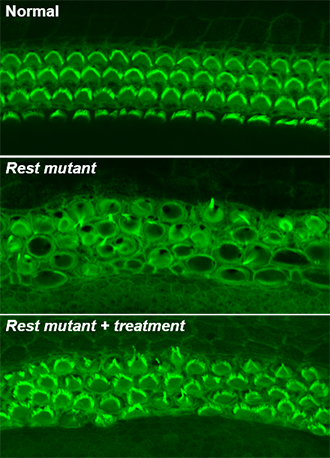
Source: Yoko Nakano, Carver College of Medicine, University of Iowa
The seed for the advance was planted a decade ago, when NIDCD researchers led by Friedman and Robert J. Morell, Ph.D., another coauthor of the current study, analyzed the genomes of members of an extended family, dubbed LMG2. Deafness is genetically dominant in the LMG2 family, meaning that a child needs to inherit only one copy of the defective gene from a parent to have progressive hearing loss.
The investigators localized the deafness-causing mutation to a region on chromosome four called DFNA27, which includes a dozen or so genes. The precise location of the mutation eluded the NIDCD team, however.
A crucial clue to explain the DFNA27 form of progressive deafness arose from later studies of the mouse RE1 Silencing Transcription Factor, or Rest, gene conducted by researchers at the University of Iowa. Botond Banfi, M.D., Ph.D. and Yoko Nakano, Ph.D., lead authors of the current study, discovered that mouse Rest is regulated through an unusual mechanism in the sensory cells of the inner ear, and this regulation is critical for hearing in mice. Because the human counterpart of the mouse Rest gene is located in the DFNA27 region, the Iowa and NIDCD researchers teamed up to reexamine the mystery of DFNA27 progressive deafness.
The coding sequence of a protein is generated from a gene by stitching together segments called exons while editing out the intervening segments. The resulting molecule serves as the template for a specific protein. Most previous studies had missed exon 4 in the Rest gene because this small exon is not edited into the Rest mRNA in most cells. The normal function of the REST protein is to shut off genes that need to be active only in a very few cell types.
When Banfi’s team deleted exon 4 of Rest in mice, inner ear hair cells died, and mice became deaf. Many genes that should have been active were shut off in hair cells prior to their death. Working together, Friedman’s and Banfi’s groups pinpointed the deafness mutation in the LMG2 family. They discovered that the mutation lies near exon 4, altering the boundaries of exon 4, and interferes with the inactivation of REST in hair cells.
“We found that incorporating exon 4 into the REST mRNA acts like a switch in sensory hair cells. It turns off REST and allows many genes to be turned on,” said Banfi. “Some of these turned-on genes are important for hair cell survival and hearing.”
The investigators used Banfi’s exon 4-deficient mice as a model for DFNA27 deafness. Since REST suppresses gene expression through a process called histone deacetylation, they wanted to see if blocking this process could reduce hearing loss. Using small-molecule drugs that inhibit this process, the investigators were able to turn off REST and partially restore hearing.
“These results demonstrate the value of studying the molecular mechanisms that underlie inherited forms of deafness,” said Andrew J. Griffith, scientific director of the NIDCD. “By following these genetic leads, we find novel and unexpected pathways that can, in cases such as this one, uncover unexpected potential treatment strategies in humans.”
This project was supported by NIDCD extramural grants (R01-DC010152 and R01-DC014953) and NIDCD Intramural Research Program funds (DC000048, DC000086 and DC000059).
About the National Institute on Deafness and Other Communication Disorders (NIDCD): NIDCD supports and conducts research and research training on the normal and disordered processes of hearing, balance, taste, smell, voice, speech, and language and provides health information, based upon scientific discovery, to the public.
About the National Institutes of Health (NIH): NIH, the nation's medical research agency, includes 27 Institutes and Centers and is a component of the U.S. Department of Health and Human Services. NIH is the primary federal agency conducting and supporting basic, clinical, and translational medical research, and is investigating the causes, treatments, and cures for both common and rare diseases.
