
Investigators at the National Institutes of Health (NIH) have uncovered a set of DNA variants, or a DNA “fingerprint,” associated with Enlarged Vestibular Aqueduct (EVA), an inherited form of hearing impairment. The variants lie near a known EVA-linked gene called SLC26A4, but their connection to EVA had evaded scientists until now. The discovery will improve methods for diagnosing EVA and may lead to new ways to restore SLC26A4 function and preserve hearing in those with the condition. The study appeared August 4, 2017, in the Journal of Medical Genetics(link is external).
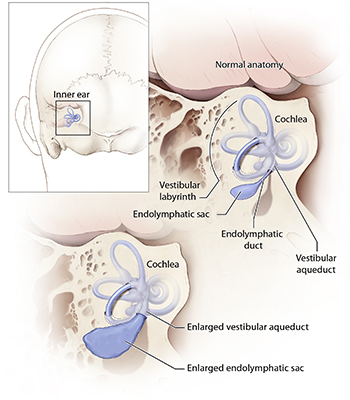
Mutations in SLC26A4 cause a hereditary hearing loss disorder characterized by enlargement of a structure in the inner ear called the vestibular aqueduct. Credit: NIH Medical Arts.
“Now that we’ve found this fingerprint—which we’ve been seeking for quite some time—we can use it to help identify the cause of hearing loss in children with EVA,” said Andrew J. Griffith, M.D., Ph.D., laboratory chief of the National Institute on Deafness and Other Communication Disorders (NIDCD) Molecular Biology and Genetics Section, who led the study. “We now have a genetic method that can inform the outcome of clinical progression and be used for testing in siblings to determine their chance of developing EVA.”
The vestibular aqueduct is a small channel that extends from the inner ear toward the brain, and serves as a conduit for the fluid that fills the inner ear. People with EVA have an enlarged channel, but scientists do not believe that the malformation itself causes hearing loss. Rather, it appears that both the structural anomaly and hearing loss are secondary to another defect.
Most children with EVA pass newborn hearing tests but begin to lose their hearing in the first few months or years of their lives. While their hearing generally declines over time, the pattern is unpredictable, with abrupt drops interspersed with episodes of brief recovery. Usually, hearing loss is the only sign of EVA, but when it is accompanied by thyroid problems the condition is known as Pendred syndrome.
The causes of EVA are complex and not fully understood, but a breakthrough came in 1997, when mutations in SLC26A4 were first linked to the condition. About half of EVA cases are caused by defects in this gene, which codes for a type of protein called an ion channel. Hearing loss in these patients likely stems from disturbances in ion balances necessary for maintaining healthy cells and transmitting sound signals to the brain. The other half of EVA patients lack evidence of SLC26A4 mutations, suggesting that other genetic or environmental factors are involved.
People with mutations in both copies of SLC26A4 typically have a more serious form of EVA. Hearing loss usually starts sooner, is more profound, occurs in both ears, and is more likely to involve the thyroid problems typical of Pendred syndrome.
As might be expected, having a single mutated copy of SLC26A4 is less harmful. Hearing loss in these individuals is not as pronounced and often only affects one ear. EVA is classified as M0, M1 or M2 based on whether it involves zero, one or two mutated copies of the SLC26A4 gene, respectively.
While people with M1 EVA were thought to have just one aberrant copy of SLC26A4, scientists were puzzled by data showing that the EVA inheritance pattern in these families is similar to that of families with M2 EVA. This observation led researchers to wonder if there was an undetected mutation in or near the “normal” SLC26A4 gene of M1 EVA patients.
To find out, Dr. Griffith’s team set out to reexamine the region of DNA that spans the seemingly normal SLC26A4 gene in M1 EVA patients from several families seen at the NIH Clinical Center. Their analysis revealed a pattern of 12 genetic variants in seven of 10 EVA patients tested. They saw the identical pattern in six unrelated Danish individuals with M1 EVA, a discovery that emerged from a collaboration with a team of Danish researchers studying EVA. These findings provided strong evidence for a causative link between the 12-variant set and M1 EVA.
Rather than being situated within SLC26A4 itself, the 12-variant fingerprint lies in front of it, an area important for controlling the gene’s activity. By altering this crucial region of DNA, the variants may dampen the SLC26A4 gene’s activity, thereby contributing to reduced SLC26A4 function. However, as M1 EVA is generally less serious than M2, the effects of the fingerprint are likely less damaging than mutations that lie within the SLC26A4 gene itself.
Additional experiments showed that the 12-variant fingerprint plays little, if any, role in people with M0 EVA. This result is consistent with other studies indicating that this form of EVA is likely caused by contributions from multiple factors unrelated to SLC26A4.
“A lot remains to be learned about what causes EVA and what the future holds for children with the disorder,” said Dr. Griffith. “But by adding to what we know about its underlying genetics, we’ve made it easier to diagnose and laid the groundwork for the development of new ways to treat it.”
In future work, Dr. Griffith plans to explore the mechanism by which the fingerprint affects SLC26A4 function, as well as the contributions made by the individual variants.
To learn more about enlarged vestibular aqueduct, visit the NIDCD EVA page.
This study was supported by the Intramural Research Program at the NIH’s NIDCD (Z01-DC000060, Z01-DC000086, Z01-DC000046, Z01-DC000039, and National Library of Medicine (Z01-LM000097).
